Hybrid peeling for fast and accurate calling, phasing, and imputation with sequence data of any coverage in pedigrees (AlphaPeel)
Peeling, concept généraux
Le peeling est une méthode dont les objectifs sont: - D’inferré des génotypes des individus - De définir les haplotypes des individus
En se basant sur le génotype de l’individu ainsi que le génotype de ces parents (néssécite sa généalogie).
le Phasing permet de ‘retrouver’ quel parent est a l’origine de l’allele a un certain locus Le peeling est “Acctuellement” insoluble avec un jeux de donnée conséquent i.e. Travailler sur l’ensemble du génome avec une grande généalogie qui fait des boucles.
le Peeling s’effectu de manière itérative. On fait un peeling up pour inférer a partir du génotype fils, a l’inverse le peeling down infére le génotype a partir du génotype ancetre.
Single‑locus peeling
Annotation: - pi (gi) : Probabilitée que l’individu i soit de génotype gi - anteriori (gi) - posteriori (gi) - penetrancei (gi)
Dérouler:
Pour affiner de plus en plus les l’inférences on fait des cycles: on propage un peeling down depuis l’ancetre le plus vieu jusqu’a inférer le déscendant le plus jeune. Puis on remonte en propagent un peeling up depuis le déscendant le plus jeune. /! on bloque le nombre de cycle maximum a 20 /!
On initialise tous les posterior (les génotype ancétre) a une constante. Puis on déroule une vague de peeling down pour mettre a jour les anterior, et on refait la mem chose en sens inversse avec le peeling up.
Parametres a calculer
- p: Fréquence de l’allele mineur
- ε: taux d’erreur de chip array
- δ: taux d’erreur de séquencage
Multi‑locus peeling
C’est un méthode qui élargie le principe du single locus peeling en prendand en compte une fonction de transmission. Cette fonction permet d’intégrer le fait que si des locus sont proche alors ils ont une grande chance d’appartenir a un haplotype identique.
Ce qui fait de cette méthode une méthode forward–backward et up-down Cette aproche fait intervenir de nouvelles choses: - tr(): Une fonction de transmission - segi,j: Un indice de segregation de l’individu i au locus j #? Segregation? - γ: le taux de recombinaison (qui sert a calculer la probabilitée de ségrégation)
Hybrid peeling
C’est une approximation computable du Multi‑locus peeling. Elle considaire que les probabilité de ségrégation entre loci proches sont égale. L’approche de la méthode est de séparer notre jeu de loci en deux jeux: A et B. avec: |A|<<|B|, A qui a les loci avec la plus grande quantitée de SNP (Ce sont nos donnée de chip Array) et B qui a les donnée du génome entier (ce sont nos donnée de séquencage).
On considére également une notion de proximité a qui est la distance relative de j a k par rapport a j+1 a j avec j et j+1 des locus de A qui englobe le locus k.
Résultats
Calling and phasing
Le hybrid peeling semble avoir de bon rendements et permet avec une bonne précision d’imputer le génotype d’individus non séquencer (ceux génotyper par chi array??) 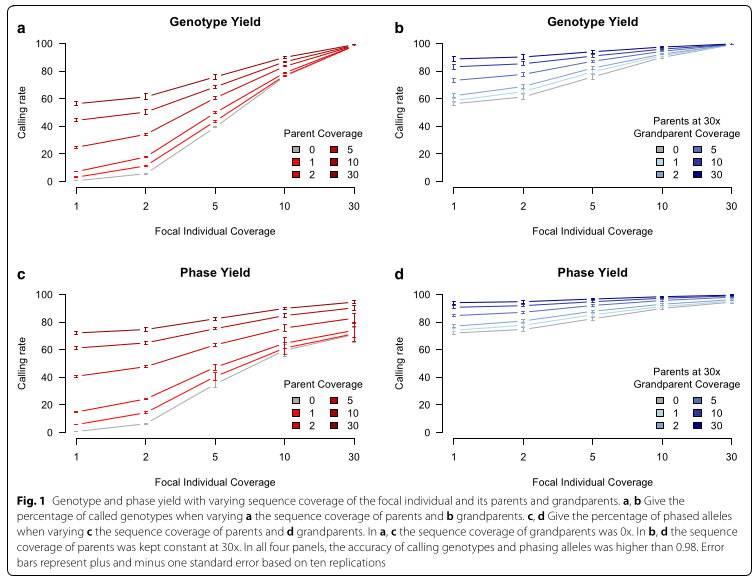
- Le calling est la détection automatique de SNP c’est une opération pas totalement précise car certaines “mutation” observé peuvent étre du a une erreur de séquencage.
- Le Yeild signifie le rendement ( ce doit étre un rapport entre le prix de génotypage et l’information obtenu)
Le gain d’information sur l’individu i nes pas très élever si le séquencage de i est élever (~12% pour du 30X) mais est plus élever si le séquencage est moins profond (~22% pour du 1X) cette augmentation d’accuracy sur le phasing se fait au détriment de la variance. cette méthode est plutot inégale.
Pour prédire la phase il est interessant de n’utiliser seulement la ‘famille proche’ pour ne pas parasiter le phasing/Genotype. Mais si on prend en compte le sequencing coverage on all ancestor #? il deviens plus interessant de travailler avec la généalogie complete.
Imputing whole‑genome sequence
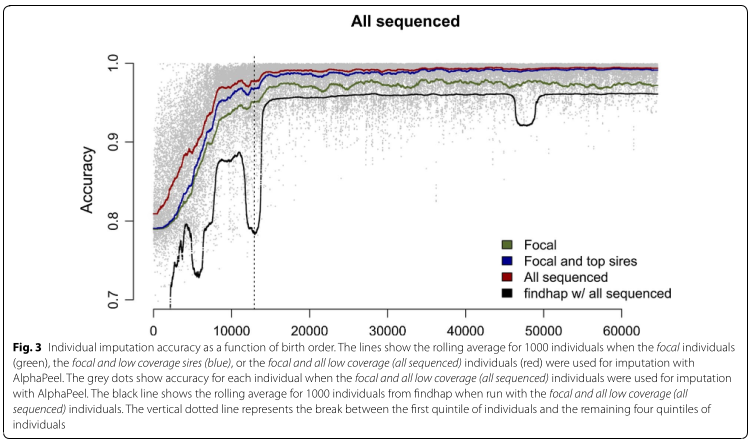
Il y a une séparation nette en therme d’accuracy entre le premier quintile et les 4 autres. Le premier quintile correspond aux ancetres, cette baisse d’accuracy est du a: - l’écart de coverage (83X de profondeur cumulé Contre 308X en moyenne) - la faible proportion d’individu avec des donnée SNP array dense
Aspect computationel
La méthode hybride est bien plus digeste que la version multi-locus (6x plus rapide, et utilise moins de memoire) aussi la méthode hybride traite les alleles indépendaments ce qui permet la parallelisation du traitement.
Discussion
Phasage et calling
L’hybrid peeling identifie les segments de chromosome partagés entre les différents parents puis propage cette information a tous les individus qui ont ce segment, mais cela est difficile quand les donnée n’ont pas une bonne couverture de la sequence. Pour palier ce probleme l’hybrid peeling marginalise au maximum les probabilitée de ségrégation #? Pour le phasing la choses la plus importante c’est la quantitée de couverture de séquences pour tous les ancetre de l’individu. Cette méthode permet de valoriser des donnée meme s’il n’existe pas de lien direct avec les individus a imputer.
Imputation
L’imputation a une meilleure précisions a la fin du pédigree. Il est plus facille d’imputer les enfants a partir des parents que l’inverse (ce phénoméne est du a la difficulté asymétrique du phasage) On obtient des résutats interessants sur les individus non-génotypé pour les 4 derniers quintile on a une précision de ~96% qui est très proche des résultats pour les individus génotypé (~98.8). En se basant uniquement sur leurs emplacement dans le pédigré et sa généalogie.
An efficient algorithm to compute the posterior genotypic distribution for every member of a pedigree without loops
Dans cet article parru 3 ans apres celui de Fernando et al. qui introduisais le Peeling. on reprnd le concept du Peeling en y intégrants les génotype, en effet dans le papier de 93 les génotypages n’existais pas on parlais alors de phénotype.
Problemes du peeling sur des grands pédigrées
Il est fréquent quand il sagit d’espece d’élevage d’avoir des boucles dans le pédigrer du a la sélection. (un géniteur génétiquement interessant peut s’accoupler dans plusieurs génération et donc parfois avec ces propre déscendants). On obtient alors des dépendance circulaire des génotype ce qui créé une explosion de la complexité car elle est exponentielle par rapport au nombre de boucles. ## Glossaire Founders: les noeuds sans parents dans le pédigrer (point terminal de la réccurence)
Résoudre l’issue 7 du projet yapp.peeling
background
L’algorythme du Peeling permet d’utiliser l’information du pedigree pour imputer des génotype. Pour ce faire le programme a besoin de lire des fichiers qui stocke des information de Pedigree et de Génotype. il est don nésséssaire de developer des fonctions capables de lire de tels fichiers. Dans le peeling itératif on a besoin d’utiliser des donnée de Pedigree. ce genre de donnée, stocker dans un fichier .ped, est présenter comme ceci:
Fichier de pedigree:
\[ \left[ \begin{array}{c|cccc} \text{ individu} \downarrow & \text{???} & \text{id} & \text{parent1} & \text{parent2} \\ \hline \text{1} &0& toto & 0 & 0 \\ \text{2} &0& tutut & 0 & 0 \\ \text{3} &0& znizni & toto & tutut\\ \end{array} \right] \]
ici les parents de \(znizni\) sont \(toto\) et \(tutut\). et \(toto\) et \(tutut\) n’ont pas de parents référencer dans le fichier. L’objectif est de créer une fonction capable de lire le fichier et de construire un graphe qui relie les parents aux enfants.
Dans le peeling itératif on a besoin d’utiliser des donnée de génotypage. ce genre de donnée est présenter comme ceci: #### Fichier génotypage: \[ \left[ \begin{array}{c|ccc} \text{} individu \downarrow & \text{id} & \text{genotype locus 1} & \text{genotype locus 2}& \text{etc..} \\ \hline \text{1} & toto & 0 & 1 &...\\ \text{2} & tutut & 2 & 0 &...\\ \text{3} & znizni & 1 & 1&...\\ \end{array} \right] \]
Dans la fonction LoadGenotypes la fonction faisais le postulat que les animaux étais identifier par des numéros et qu’il n’y avais pas de ‘trous’ dans la liste ( individus numéroté de 1 a n avec n le nombre d’individu) bien qu’ils peuvent etre dans le désordre:
return genotypes_array[IDs - 1, :]Issue
Cette approche génére un probleme si le format n’est pas respecter,Par exemple quand l’IDs d’un individu n’est pas un entier. Et cela entraine des erreurs.
L’objecif serais de pouvoir utiliser le numéro officiel (ou autre type d’identifiant) de l’animal comme identifiant et d’avoir en suite la référence de son génome dans la np.array.
Une solution pour ça est de créer un dictionnaire qui prendrais en clé un String et qui renverai un indexe de la np.array des génotype. Et ainsi séparer les Identifiants des Indexes.
Une autre solution est, lors de la lecture du pedigree, de générer un indexe et de le stocker dans chaque PedNode. l’avantage est que’il n’y a pas besoin de créer une nouvelle structure de donnée.
/! L’identifiant de l’animal doit étre le même que dans le fichier de pedigree /! . c’est lors de la lecture du pedigree qu’on créer les PedNodes, j’ai donc modifier le constructeur de PedNodePeel pour pouvoir implementer la propiété idx. Ainsi lors de la création des individus un indexe leur ai associe.
Aussi j’ai du réécrire la fonction add_indiv pour qu’elle prenne en compte la syntaxe du nouveau constructeur de PedNodePeel :
# Overwrite
def add_indiv(self, indiv):
"""Add a new guy to the pedigree"""
if indiv in self.nodes:
logger.debug(f"{indiv} is already there")
else:
self.nodes[indiv] = self.node_class(indiv, self.__current_idx)
self.__current_idx += 1Un parametre current_idx a etre créer dans la classe PedigreePeel pour tenir a jour le compteur d’indexes remarque: on aurais pu calculer l’indexe a partir de la taille du pédigree. /! Lors de la suppession d’un individus les indexes serait décalé/! .
Test
Une fois les modifications faites, il faut tester si les changement supporte bien le “nouveau” format de donnée. Pour ce faire j’ai modifier un fichier .ped en faisant trois choses:
- modifier les nom de quelques individus pour qu’il ne soit plus des nombres
- supprimer une ligne de géniteur (individu 14)
- ajouter un pere non présent dans le pédigrer
# Pedigee_alterer.ped
0 1 0 0 1 -9
0 2 0 0 2 -9
0 3 1 2 1 -9
0 4 0 0 2 -9
0 5 0 0 1 -9
0 6 0 0 2 -9
0 7 0 0 1 -9
0 8 0 0 2 -9
0 9 5 6 2 -9
0 10 3 4 1 -9
0 11 0 0 2 -9
0 francis 3 4 1 -9
0 13 7 8 2 -9
0 15 10 9 1 -9
0 16 10 9 1 -9
0 17 ici 11 1 -9
0 18 francis 11 1 -9
0 19 francis 13 1 -9
0 20 francis 13 1 -9
0 21 0 13 1 -9
0 zog 0 13 1 -9
0 23 0 0 2 -9
0 toto zog 23 1 -9
0 25 zog 23 1 -9Résultats
Pour le pedigree le résultat est satisfaisant on est bien arriver a faire coresspondre l’indexe avec le nom de l’individu.
import yapp.peeling as pl
pedfile = "Pedigee_alterer.ped"
ped = pl.PedigreePeel.from_fam_file(pedfile)
[[ped.nodes[i].indiv,ped.nodes[i].idx] for i in ped.nodes]
#Out[1]:
[['1', 0],
['2', 1],
['3', 2],
['4', 3],
['5', 4],
['6', 5],
['7', 6],
['8', 7],
['9', 8],
['10', 9],
['11', 10],
['francis', 11],
['13', 12],
['15', 13],
['16', 14],
['17', 15],
['ici', 16],
['18', 17],
['19', 18],
['20', 19],
['21', 20],
['zog', 21],
['23', 22],
['toto', 23],
['25', 24]]ici est créer juste apres 17 car il est référencer comme parent mais n’a pas encore été traité comme individu.
Concernant le génotype, plusieurs fonction permetais de lire les fichiers de génotypage. Deux fonctions a modifier: - LoadGenotype qui lit le génotypage a partir d’un .csv - ReadVCF qui lui utilise un fichier .vcf
Apres modification voici Load Genotype
def LoadGenotypes(file: str, ped: PedigreePeel, sep: str = "\t", unknowGenotype=3) -> np.ndarray:
# Read the CSV file into a list of lists
# Init np.full (nInd,nSNP) -> -9 ou 3
# nSNP can be found by counting the ncol on the first csv
# then fill geno with geno[ped.nodes[row[0]].idx,...] = row[1:]
with open(file, newline="") as csvfile:
reader = csv.reader(csvfile, delimiter=sep)
# Read the first row to initialise np array
firstrow = next(reader)
# Create np.array (nInd, nSNP) full of "unknowGenotype" int: 3 by default
genotypes_array = np.full((len(ped.nodes), (len(firstrow) - 1)), unknowGenotype)
genotypes_array[ped.nodes[firstrow[0]].idx, ...] = firstrow[1:]
for row in reader:
try:
genotypes_array[ped.nodes[row[0]].idx, ...] = row[1:]
except KeyError:
print(row[0], "is not in the pedigree: individual ignored")
return genotypes_arrayon peut noter la présence du pedigree dans les parametres, néssésaire pour faire la corresspondance entre les indexe (stocker dans les PedNodePeel) et les génotypes. Pour tester les modifications et la bonne correspondance entre individu et génotype j’ai créer un fichier “genotype_fernando_realt_shuf.csv” ou j’ai intervertie des lignes du fichier de génotypage et j’ai également ajouter une ligne avec un individu nomé “l’intru” qui n’est pas présent dans le pedigree pour tester ce cas.
genfile_shuffled = "genotype_fernando_realt_shuf.csv"
geno_ctrl = pl.LoadGenotypes(genfile,ped,',')
geno_shuff = pl.LoadGenotypes(genfile_shuffled,ped,',')
geno_ctrl == geno_shuff
#Out[1]:
#l'intru is not in the pedigree: individual ignored
array([[ True, True, True, True, True, True, True, True, True, True],
[ True, True, True, True, True, True, True, True, True, True],
[ True, True, True, True, True, True, True, True, True, True],
[ True, True, True, True, True, True, True, True, True, True],
[ True, True, True, True, True, True, True, True, True, True],
[ True, True, True, True, True, True, True, True, True, True],
[ True, True, True, True, True, True, True, True, True, True],
[ True, True, True, True, True, True, True, True, True, True],
[ True, True, True, True, True, True, True, True, True, True],
[ True, True, True, True, True, True, True, True, True, True],
[ True, True, True, True, True, True, True, True, True, True],
[ True, True, True, True, True, True, True, True, True, True],
[ True, True, True, True, True, True, True, True, True, True],
[ True, True, True, True, True, True, True, True, True, True],
[ True, True, True, True, True, True, True, True, True, True],
[ True, True, True, True, True, True, True, True, True, True],
[ True, True, True, True, True, True, True, True, True, True],
[ True, True, True, True, True, True, True, True, True, True],
[ True, True, True, True, True, True, True, True, True, True],
[ True, True, True, True, True, True, True, True, True, True],
[ True, True, True, True, True, True, True, True, True, True],
[ True, True, True, True, True, True, True, True, True, True],
[ True, True, True, True, True, True, True, True, True, True],
[ True, True, True, True, True, True, True, True, True, True],
[ True, True, True, True, True, True, True, True, True, True]])Puis pour ReadVCF
def ReadVCF(vcfile: str, ped, mode: str = "likelihood", baseValue: float = 0.0) -> np.ndarray:
samples = list(ped.nodes.keys()) # Try to remove list() when it's done
vcfdat = vcf.vcf2fph(vcfile, samples=samples, mode=mode)
assert mode in [
"likelihood",
"genotype",
], "mode can only be likelihood or genotype."
# vcfdat is organized per chromosome, so we need to merge them
# Pre allocation table
nInd, nLoci = len(samples), sum(
[len(vcfdat["variants"][key]) for key in vcfdat["variants"].keys()]
)
if mode == "likelihood":
genotypes = np.full((nLoci, 3, nInd), baseValue, dtype=np.float32)
else:
genotypes = np.full((nLoci, nInd), baseValue, dtype=np.int8)
for chr in vcfdat["regions"]:
for node in ped:
try:
idx = node.idx
genotypes[..., idx] = vcfdat["data"][chr][node.indiv]
except KeyError:
logger.warning(f"Individual {node.indiv} missing in vcf.")
return genotypesChangement de direction
Apres l’entretient hebdomadaire il c’est avéré que le stockage de l’index dans le PedNodePeel posé un problème sémentique: En effet, l’indexe est relatif a une collection de PedNode et pas un seul PedNode. Donc la présence de l’attribut “idx” dans PedNode ne fait pas sens.
Donc la solution de l’utilisation d’un dictionnaire “individus-indexe” semble plus adéquate.
Bug lors du test de l’iterative peeling
En vue de tester les modifications de code j’ai tenter de lancer l’iterative peeling avec mon fichier de genotypage altérer mais le read csv pose probleme , le programme considére qu’il existe 4 génotype possible alors qu’il en existe 3.
Le probléme venais de la gestion du génotype manquant: dans le fichier un 3 encodé un génotype inconnu mais dans le programme c’est -1 qui es considérer comme une ‘missing value’.
Calcul de PopFreq
Pour effectuer le Peeling il faut connaitre les valeurs d’anterieur des fondateur. vue que les fondateurs ont par définition pas de parents présent dans le génotype, on utilise alors les fréquence des génotypes calculé sur l’ensembles des fondateur comme valeur des anterieur des fondateur. Pour pouvoir obtenir cette fréquence de génotypes on doit calculer la \(MAF\) (minor allele frequency). La MAF peut se calculer comme ceci \(MAF = {1\over 2}P(Aa)+P(aa)\) Sauf qu’il est impossible de trouver la vrais probabilité d’un génotype. Mais le peeling permet d’avoir \(P(g|D)\) avec \(g\) un génotype et \(D\) les génotypages (les données observées), il est impossible de calculer la \(MAF\) mais grace au peeling on peut calculer un estimateur: \(\widehat{MAF}\).
\[\widehat{MAF} = {1\over N}\sum_{i}{0.5 \widehat{P_i}(Aa) + 1\widehat{P_i}(aa)} \]
Avec \(N\) le nombre total de fondateurs et \(\widehat{P_i}(AA)\) la probabilitée estimée que l’individu i soit de génotype AA, dans le cadre du Peeling cette valeur est la probabilitée d’observé le génotype \(AA\) sachants les donnée observé \(D\). \(\widehat{P_i}(AA) = P_i(AA|D)\)
Une fois la \(\widehat{MAF}\) on peut alors calculer une estimation distribution des génotypes (Hardy-Weinberg): \[ P(G_i = g \mid p ) = \begin{cases} (1-p)^2 \\ 2p(1-p) \\ p^2 \end{cases} \] Avec \(p\) = \(\widehat{MAF}\)
Tests
L’objectif ici est de pouvoir calculer la fréquence allélique des fondateurs a chaque loci, pour pouvoir simuler leurs antérieurs. cette valeur estimée de \(\widehat{MAF}\) est sensé, au cour du peeling, se rapprocher de la valeur exacte de \(MAF\).
Pour pouvoir tester si \(\widehat{MAF}\) se rapproche de la valeur exacte \(MAF\), le plus simple est de travailler avec un jeux de donnée simulé. En effet un jeux de donnée simulé nous permet d’avoir la valeur exacte de \(MAF\), ainsi on peut comparer l’évolution de \(\widehat{MAF}\) par rapport a \(MAF\)
Présentation du jeux de donnée de test
Ici nous utiliseron un jeux de donné avec un pédigré de 550 individus, 11 générations et 50 individus par génération. Ce qui fait donc 50 fondateurs (individus de premieres génération). Le jeux de donnée contient également des donnée de génotypage, un vrai génotype ainsi qu’un fichier de génotypage simulée avec une likelyhood, on a aussi acces au vrais fréquences alléliques des fondateurs.
On peut donc tester plusieurs choses:
- si on retombe bien sur la \(MAF\) quand on la calcule a partir des vrais génotype
- si la \(\widehat{MAF}\) évolue au fur et a mesure du peeling
- regarder l’évolution de l’erreur quadratique \(MSE\) entre \(MAF\) et \(\widehat{MAF}\)
Tests
Test de la formule de calcul de la MAF:
pedfile = "genotype.ped"
genfile = "genotype_falseGL.vcf.gz"
with open("founders_frequencies.pickle","rb") as picfile:
fndrFreq = pickle.load(picfile)[0]
ped = pl.PedigreePeel.from_fam_file(pedfile)
samples = [ped.nodes[i].indiv for i in ped.nodes.keys()]
idx_dict = {}
for index, value in enumerate(samples):
idx_dict[value] = index
trueGeno = pl.LoadGenotypes("genotype.txt",idx_dict)
#turn (550, 1000) shape into (1000, 3, 550)
trueGeno = np.eye(3)[trueGeno] #<-(550,1000,3), one hot encoding
trueGeno = np.transpose(trueGeno,(1,2,0))#<-(1000, 3, 550)
fdridx = [idx_dict[i.indiv] for i in ped.founders]
trueGeno = trueGeno[...,fdridx] #filter on founders
#____MAF Computation____#
MAF_from_trueGeno = trueGeno * np.array([0, 0.5, 1])[None, :, None]
MAF_from_trueGeno = np.sum(MAF_from_trueGeno, axis=(1, 2)) / trueGeno.shape[-1]
#____Test____#
test = np.array(list(fndrFreq.values())== MAF_from_trueGeno)
if np.prod(test) != 0: #si les deux lectures matches ( en booléen * = And , + = Or)
print ("test du calcul de MAF : réussite")
else:
print ("test du calcul de MAF : échec")test du calcul de MAF : réussiteTest de l’évolutuion de l’estimation de la \(\widehat{MAF}\) avec \(MSE\):
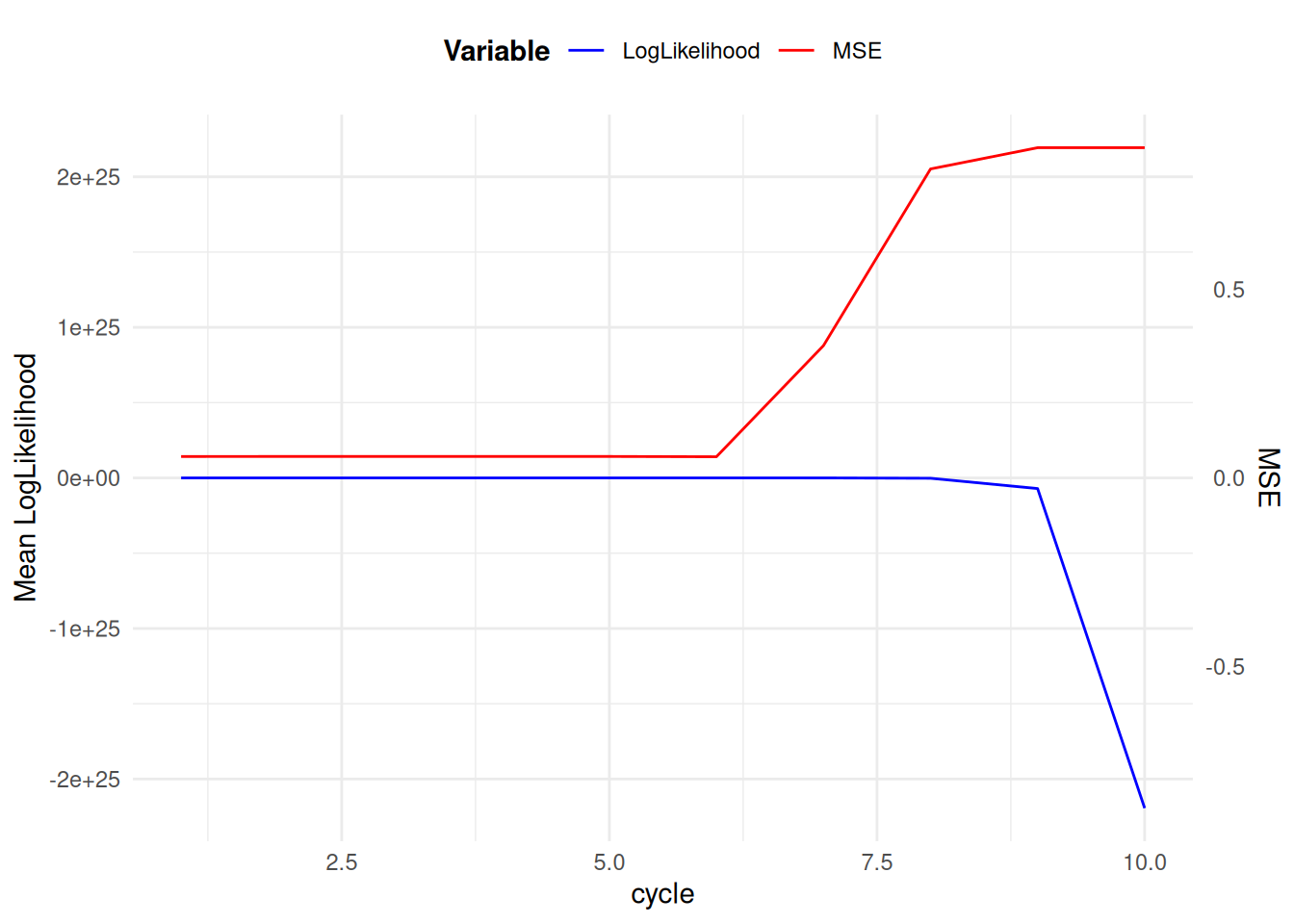
Le Bug
Au vue des résultat une erreur c’est glissé dans le code nous donnants des résultats abérant de vraisemblance. en effet il est étonnant d’obtenir une log vraisemblance a -2 000 000 000 dès la premiere itération, de plus la qualitée du Peeling est sensé améliorer la vraisemblance sauf qu’ici elle diminu.
Trouver le bug
Une telle erreur est difficile a identifier car elle ne résulte pas en l’arret du script, elle s’imisse dans les résultats et n’est réelement visible
symptome de l’erreur
la logvraisemblance totale est mauvaise et la premiere choses que nous avons fait c’est dès la premiere itération regarder si les valeurs pour la calculer sont aussi bizarres: \(L = \sum_{u_i}{\textcolor{red}{anterior}_i(u_i)\textcolor{green}{penetrance}_i(u_i) \prod_{j \in S_i}} \textcolor{blue}{posterior}_{ij}(u_i)\)
On a donc regarder les valeurs de l’\(\textcolor{red}{anterior}\) qui ne semblais pas abérantes, idem pour la \(\textcolor{green}{penetrance}\), mais quant aux posterieurs les on valeurs extrèmes ( -10 Milliards), au premier abord on pensais que le problème venais de la fonctions qui calculer ce \(\textcolor{blue}{posterior}\).
Mais on c’est ensuite rendu compte que ce qui posait probléme était en fait l’addition consécutive de valeurs, en effet: lors du peel UP on parcoure le pédigrée génération par génération par ordre décroissant. Donc les premiers calculs de posteriors se base sur les antériors et pénétrances mais pas les posteriors car la derniere génération n’a pas de descandants. Sur notre jeu de donnée on obtenais des valeurs d’anteriors a -800 pour les dernières générations, et vue qu’on additionne les valeurs anterior pour calculer les posterior on fini par “empiler” les valeurs et obtenir des valeurs extrèmes.
Pour palier ce problème, qui fini par etre un probleme computationnel car on ne peut pas faire de calcul précis avec des nombres trop extreme, il faut scalé les données.
Scaling
Démonstration de la Scalabilitée du peeling
On sait que la formule du Peeling permet d’obtenir
\(Pr(u_i|y) \propto a_i(u_i) g(y_i|u_i) \prod_j p_{ij}(u_i)\)
Or scaler les termes du peeling reviens a dire que: \[ \begin{cases} a_i(u_i)=C_i \centerdot a_i^* (u_i) \\ g_i(y_i|u_i)=E_i \centerdot g_i^*(y_i|u_i) \\ p_{ij}(u_i) = D_{ij} \centerdot p_{ij}^*(u_i) \end{cases} \] Avec \(C_i\), \(E_i\), \(D_ij\) des constantes.
Probabilitée a posteriorie
En faisant l’hypothése que les thermes \(a_i(u_i)\), \(g_i(y_i|u_i)\) et \(p_{ij}(u_i)\) sont scalable on a donc:
\(Pr(u_i|y) \propto a_i(u_i) g(y_i|u_i) \prod_j p_{ij}(u_i)\)
\(Pr(u_i|y) \propto \textcolor{darkred}{C_i}a_i^*(u_i) \textcolor{darkred}{E_i}g^*(y_i|u_i) \prod_j \textcolor{darkred}{D_{ij}} p_{ij}^*(u_i)\)
\(Pr(u_i|y) \propto \textcolor{darkred}{C_i E_i \prod_j D_{ij}} \centerdot a_i^*(u_i) g^*(y_i|u_i) p_{ij}^*(u_i)\)
\(Pr(u_i|y) \propto a_i^*(u_i) g^*(y_i|u_i) p_{ij}^*(u_i)\)
Donc la relation de proportionnalitée est conservé avec des donnée scalées.
Anterior
\[ \begin{align} a_i(u_i) =\sum_{u_m}\Bigg\{ a_m(u_m)g(y_m|u_m)\prod_{\stackrel{j\in S_m}{j\ne f}}p_{mj}(u_m) \times \\ \sum_{u_f}\Bigg\{a_f(u_f)g(y_f|u_f)\prod_{j'} p_{fj'}(u_f)\times tr(u_i|um,uf) \times \\ \prod_{j"}\Bigg[\sum_{u_{j"}}tr(u_{j"}|u_m ,u_f)g(y_{j"}|u_{j"})\prod_{k}p_{kj"}(u_k)\Bigg]\Bigg\} \Bigg\} \end{align} \]
Or d’après notre hypothése de scalabilitée on a:
\[ \begin{align} a_i(u_i) =\sum_{u_m}\Bigg\{ \textcolor{darkred}{C_m}a_m(u_m)\textcolor{darkred}{E_m}g(y_m|u_m)\prod_j \textcolor{darkred}{D_{mj}} p_{mj}(u_m) \times \\ \sum_{u_f}\Bigg\{\textcolor{darkred}{C_f}a_f(u_f)\textcolor{darkred}{E_f}g(y_f|u_f)\prod_{j'} \textcolor{darkred}{D_{fj'}} p_{fj'}(u_f)\times tr(u_i|um,uf) \times \\ \prod_{j"}\Bigg[\sum_{u_{j"}}tr(u_{j"}|u_m ,u_f) \textcolor{darkred}{E_{j"}}g(y_{j"}|u_{j"})\prod_{k}\textcolor{darkred}{D_{kj"}}p_{kj"}(u_k)\Bigg]\Bigg\} \Bigg\} \end{align} \] \[ \begin{align} a_i(u_i) =\textcolor{darkred}{C_m C_f E_m E_f \prod_j D_{mj} \prod_{j'} D_{fj'} \prod_{j"} E_{j"} \prod_{k} D_{kj"} }\sum_{u_m}\Bigg\{ a_m^*(u_m)g^*(y_m|u_m)\prod_j p_{mj}^*(u_m) \times \\ \sum_{u_f}\Bigg\{a_f^*(u_f)g^*(y_f|u_f)\prod_{j'} p_{fj'}^*(u_f)\times tr(u_i|um,uf) \times \\ \prod_{j"}\Bigg[\sum_{u_{j"}}tr(u_{j"}|u_m ,u_f) g^*(y_{j"}|u_{j"})\prod_{k} p_{kj"}^*(u_k)\Bigg]\Bigg\} \Bigg\} \end{align} \]
\[ a_i(u_i) = \textcolor{darkred}{C_m C_f E_m E_f \prod_j D_{mj} \prod_{j'} D_{fj'} \prod_{j"}\Bigg[ E_{j"} \prod_{k} D_{kj"}\Bigg] } \times \textcolor{green}{\tilde{a}_i(u_i) } \]
Donc on a bien la fonction \(anterior\) qui s’écrit sous la forme:
\[
a_i(u_i) = C_i \times a_i^*(u_i)
\] avec ici \(a_i^*(u_i) = \tilde{a}_i(u_i) \times w_i\) tel que \(w_i = max_{u_i}(a_i(u_i))\)
Posterior
\[ p_{ij}(u_i) = \sum_{u_j}\Bigg[ a_j(u_j)g(j_i|u_j) \prod_k p_{jk}(u_j)\times \prod_{k'}\Big[\sum_{u_k'}tr(u_{k'}|u_i ,u_j)g(y_{k'}|u_{k'}) \prod_l p_{k'l}(u_{k'})\Big]\Bigg] \]
Or, d’après l’hypothése de scalabilitée on a:
\[ p_{ij}(u_i) = \sum_{u_j}\Bigg[ \textcolor{darkred}{C_i\centerdot} a_j^*(u_j)\textcolor{darkred}{E_i \centerdot} g^*(j_i|u_j) \prod_k \textcolor{darkred}{D_{jk}\centerdot}p_{jk}^*(u_j)\times \prod_{k'}\Big[\sum_{u_k'}tr(u_{k'}|u_i ,u_j)\textcolor{darkred}{E_k' \centerdot}g^*(y_{k'}|u_{k'}) \prod_l \textcolor{darkred}{D_{k'l} \centerdot}p_{k'l}^*(u_{k'})\Big]\Bigg] \]
\[ p_{ij}(u_i) = \textcolor{darkred}{C_i E_i \prod_k D_{jk} \prod_{k'}\big[ E_k' \prod_l D_{k'l} \big]}\times \sum_{u_j}\Bigg[ a_j^*(u_j) g^*(j_i|u_j) \prod_k p_{jk}^*(u_j)\times \prod_{k'}\Big[\sum_{u_k'}tr(u_{k'}|u_i ,u_j) g^*(y_{k'}|u_{k'}) \prod_l p_{k'l}^*(u_{k'})\Big]\Bigg] \]
\[ p_{ij}(u_i) = \textcolor{darkred}{C_i E_i \prod_k D_{jk} \prod_{k'}\big[ E_k' \prod_l D_{k'l} \big]}\times \textcolor{green}{\tilde{p}_{ij}(u_i)} \]
Donc on a bien la fonction \(posterior\) qui s’écrit sous la forme \[ p_{ij}(u_i) = D_{ij} \times p_{ij}^*(u_i) \] avec ici \(p_{ij}^*(u_i) = \tilde{p}_{ij}(u_i) \times w_i\) tel que \(w_i = max_{u_i}(p_{ij}(u_i))\)
Conclusion
Donc vu qu’il est possible d’exprimer chaque termes de la formule sous une forme scalé. Alors il est possible de travailler avec des données scalées sans altérer les résultats.
Pruning
Contexte
Dès lors ce qu’on manipule des réseaux de grande taille il arrive souvent de ne pas pouvoir valorisé toute l’information. par exemple, dans le cadre du peeling, si un sous-graphe entier du pédigrée n’est pas génotypée, alors elle n’apporte aucune information génétique, de plus l’imputation de génotype de ces individus risque d’étre très mauvaise et, par conséquent, de faire baisser la vraisemblance globale du modele. Pour pallié ce probleme un pédigré doit étre “Pruné”, cette étape permet de se séparer des “branches” jugé inutile, ou du moins inutilisable.
L’étape de pruning est “Algorithme dépendante” c’est a dire que pour un meme pédigrée le pruning peut étre différent en fonction de l’algorithme que l’on veux lancer. Le Pruning est donc une étape du developpement a part entière. Cela dit il n’existe pas de “Pruning parfait”, une des principale utilitée du pruning est de réduire le jeu de donnée pour gagner en temps de calcul, ce qui fait que parfois on préferera supprimer un individu qui pourrait apporté de l’information pour gagner en temp de calcul.
Dans le Peeling, que doit-on pruné?
La question ici est surtout de savoir quels individus non génotyper on veux garder. Le Peeling se base sur l’information génétique de l’individus, de ses parents et de ces enfants. risque d’étre difficile a imputé. Mais aussi si un individus non génotypé ne posséde aucun déscendant génotypé alors il ne produit aucune information exploitable. (imputer un génotype reviendarait a faire un tirage quasi aléatoire dans le génotype des parents)
Algorithme de pruning
L’algorithme de pruning effectu un parcour de pédigrée génératioin par génération, en partant de la derniere génération, cela permet de lire chaque individus avant ses parents. puis on associe a chaque individu une distance, cette valeur est la distance au déscendent génotypé le plus proche. le calcul de distance ce fait comme suit: \(\text{soit g un individu génotypé et k un individu non génotypé on a: }\)
\[ \newline Dist_g = 0 | Dist_k = min(Dist_{Children_k})+1 \]
Ensuite il suffit d’appliquer un seuil sur la distance pour supprimer les individus qui sont a plus de \(x\) individus d’un individu génotypé.
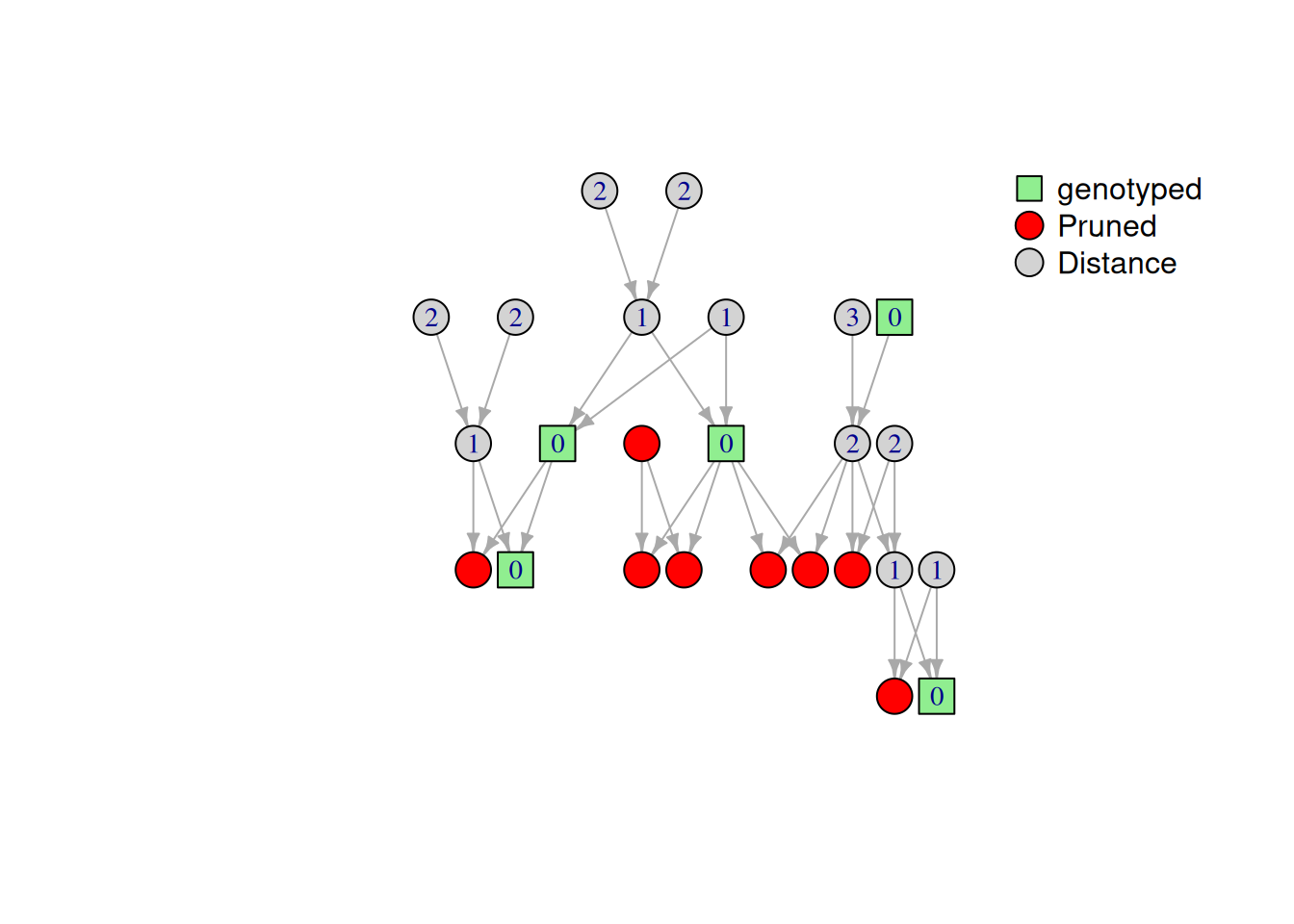
Algorithme de pruning v2
C’est l’amélioration de l’algorithme de pruning, Il calcule une distance au déscendant génotypé le plus proche, mais aussi a l’ancetre génotypé le plus proche. si la distance est inférieure a un seuil alors on ne garde l’individus que si ces déscendants directs sont génotypé et assez nombreux (>10)
Autres mesures
En plus de celle calculé dans l’algorithme du pruning v1, cet algorithme calcule une autre mesure de distance: la distance a l’ancetre génotypé le plus proche. La néssécité d’une telle mesure viens de la conception de l’algorithme du peeling, en effet la formule du peeling se base en partie sur l’information génétique apporté par les parents d’un individu. Ce qui signifie qu’il est très dur d’imputer un individu en ayant uniquement l’information génétique de ses déscendants, sauf si l’individu posséde un grand nombre d’enfants génotypé. Si c’est le cas, il est alors possible d’imputer le génotype du parent a partir de celuis de ces enfants.
L’algorithme de prunning v2 vas prendre en compte ces trois information; Distance a l’ancetre génotypé, distance au déscendant génotypé et nombre d’enfants directs génotypé , afin de “pruné” au mieux notre pédigée.
Concrétement
En l’état l’algorithme de pruning se divise en deux partie
- une partie de calcul des distance qui contient deux parcour du pédigree
- une partie qui vas filtrer et supprimer les noeuds indésirables dans le pédigree
Calul des distance
les mesures de distances sont stocker dans un default dict qui est une structure de donnée semblable a un dictionnaire mais qui renvoi une mesure par défaut si la clé n’est pas présente dans le dictionnaire. Ici nos dictionnaires de distances auront une valeur par défauts a infini, et prendrons en entrée un identifiants d’individus. ex: \(distDict['tutut'] \rightarrow 2\). nous alons utiliser deux dictionnaires de distances distincs:
le premier, distDict_gd stocke les distances des individus a leur descendant génotypé le plus proche. \(\text{soit g un individu génotypé et k un individu non génotypé, les distances sont calculer comme suit: }\) \[ \newline Dist_g = 0 | Dist_k = min(Dist_{Children_k})+1 \]
le second, distDict_ga stocke les distances des individus a leur ancetre génotypé le plus proche. \(\text{soit g un individu génotypé et k un individu non génotypé, les distances sont calculer comme suit: }\) \[ \newline Dist_g = 0 | Dist_k = min(Dist(Mother_k), Dist(Father_k))+1 \]
Filtre et suppression
la seconde partie de l’algorithme est l’application des filtre, elle parcoure tous les individus du pédigree et pour chaqu’un vérifie si des conditions logiques sont respecté. il y a deux conditions a respecter: $ $
$ $
Il est important de noté qu’un individu génotypé a une distance à un ancetre génotypé égale a zéro (idem pour la distance a un déscendant génotypé).
ped = pl.PedigreePeel.from_fam_file(pedfile)
samples = [ped.nodes[i].indiv for i in ped.nodes.keys()]
samples = samples[:175]
samples = samples[125:]
def prunning_alt(ped, gSamples, filter: int = np.inf):
allSamples = list(ped.nodes.keys())
distDict_gd = defaultdict( # gd for genotyped descendant
lambda: np.inf
) # initialise distance from a genotyped indiv to +inf
distDict_ga = defaultdict( # gd for genotyped ancestor
lambda: np.inf
) # initialise distance from a genotyped indiv to +inf
# set genotyped individuals distance to 0
for indiv in gSamples:
distDict_gd[indiv] = 0
distDict_ga[indiv] = 0
sortedPedNodes = sorted(list(ped.nodes.values()), key=lambda x: x.gen, reverse=True)
# crossing pedgraph generation by generation. First time we calculate distances
for i in sortedPedNodes:
indiv = i.indiv
if distDict_gd[indiv] != 0:
if i.children != []:
distDict_gd[indiv] = min([distDict_gd[c.indiv] for c in i.children]) + 1
sortedPedNodes = sorted(
list(ped.nodes.values()), key=lambda x: x.gen, reverse=False
)
# crossing pedgraph generation by generation. First time we calculate distances
for i in sortedPedNodes:
indiv = i.indiv
if distDict_gd[indiv] != 0:
if i.mother: # mean Father isn't None too
distDict_ga[indiv] = (
min([distDict_ga[i.mother.indiv], distDict_ga[i.father.indiv]]) + 1
)
# Removing individuals with a distance above filter
removed_cpt = 0
removed_indiv = []
for i in ped:
indiv = i.indiv
if distDict_ga[indiv] >= filter or distDict_gd[indiv] >= filter:
if len(i.children) < 10:
removed_indiv.append(ped.del_indiv(indiv))
removed_cpt += 1
else:
x = [j for j in i.children if distDict_gd[j.indiv] == 0 ]
# we dont prune individuals with at least 10 genotyped childrens
if len(x)< 10:
removed_indiv.append(ped.del_indiv(indiv))
removed_cpt += 1
print("Pruning done:", removed_cpt, "nodes removed")
return distDict_gd,distDict_ga
(distDict_gd,distDict_ga) = prunning_alt(ped,samples,3)Simulation de “vrai” penetrance:
Estimate \(P(Y|G)\) from Binomial allele sampling
- \(G\) a genotype array of shape \((L\times N)\) where \(L\) is the number of loci and \(N\) a set of samples.
- \(D \sim \text{Poisson}(D_{\text{moy}})\) where \(D\) is the sequencing depth at specific locus \(l\) for a sample \(n\) and \(D_{\text{moy}}\) the average sequencing depth for the whole experiment.
- \(R_1 \sim \text{Binomial}(D,P = f(G,\epsilon))\) with:
- \(\epsilon \in [0,1]\)
- \(f(G,\epsilon) = \begin{cases} 0/0: & \epsilon \\ 0/1: & \frac{1}{2} \\ 1/1: & 1- \epsilon \end{cases}\)
- From \(f(k) = \binom{n}{k} p^k (1-p)^{n-k}\):
- \(P(Y = \{D,R_1\}|G \in \{0,1,2\}) \begin{cases} 0: & \epsilon^{R_1} \quad (1-\epsilon^{D - R_1}) \\ 1: & \frac{1}{2}^D \\ 2: & (1- \epsilon)^{R_1} \quad \epsilon^{D - R_1} \end{cases}\)
- \(log P(Y = \{D,R_1\}|G \in \{0,1,2\}) \begin{cases} 0: & R_1 \log (\epsilon) + (D-R_1) \log (1-\epsilon) \\ 1: & \log \frac{1}{2} \\ 2: & \log (1- \epsilon) + (D - R_1) \log \epsilon{} \end{cases}\)
from scipy import stats
import numpy as np
random_state = 43
N, L, Dm, epsilon = 10, 1000, 5, 0.01
G = np.random.choice((0,1,2), size = (L,N), replace=True)
D = stats.poisson.rvs(Dm,size = (L,N), random_state = random_state)
pf = np.array((epsilon,0.5,1-epsilon))
pf_G = pf[G.ravel()].reshape((L,N))
R1 = stats.binom.rvs(D,pf_G, random_state = random_state)
penetrance = np.stack([
R1 * np.log(epsilon) + (D-R1) * np.log(1-epsilon),
np.full((L,N),np.log(0.5)),
np.log(1-epsilon) + (D-R1) * np.log(epsilon)
],axis=-1)
penetrance = np.einsum("lnu->lun",penetrance)