A beginner’s guide to low-coverage whole genome sequencing for population genomics
Contexte
Bien que le séquencage coute de moins en moins cher, Des choix subsiste quant a la profondeur, la couverture et le nombre d’individus a séquencer, on a donc un curseur a 3 dimention. Le low-pass permetrait d’obtimiser ce problème. Le low-pass (lcWGS) est une méthode économique de séquencage de génome complet chez des population.
Plusieurs problemes avec les méthodes classiques RAD-seq et Pool-seq: - une approche comme RAD-seq ne permet pas d’avoir l’information sur tous le génome, ou alors le processus serait hors de prix. - une approche comme Pool-seq est moins couteuse et efficasse si la population est assez grande mais l’information génétique individuelle est perdu.
la stratégie de low-coverage est de diffuser l’information a travers tous le génome de pleins d’individus “barcodé”. Ce qui entraine une diminution drastique de la profondeur de séquencage pour chaque individu, et donc la précision des génotypage individuels. mais ce qui augmente l’amplitude de la couverture de séquencage. 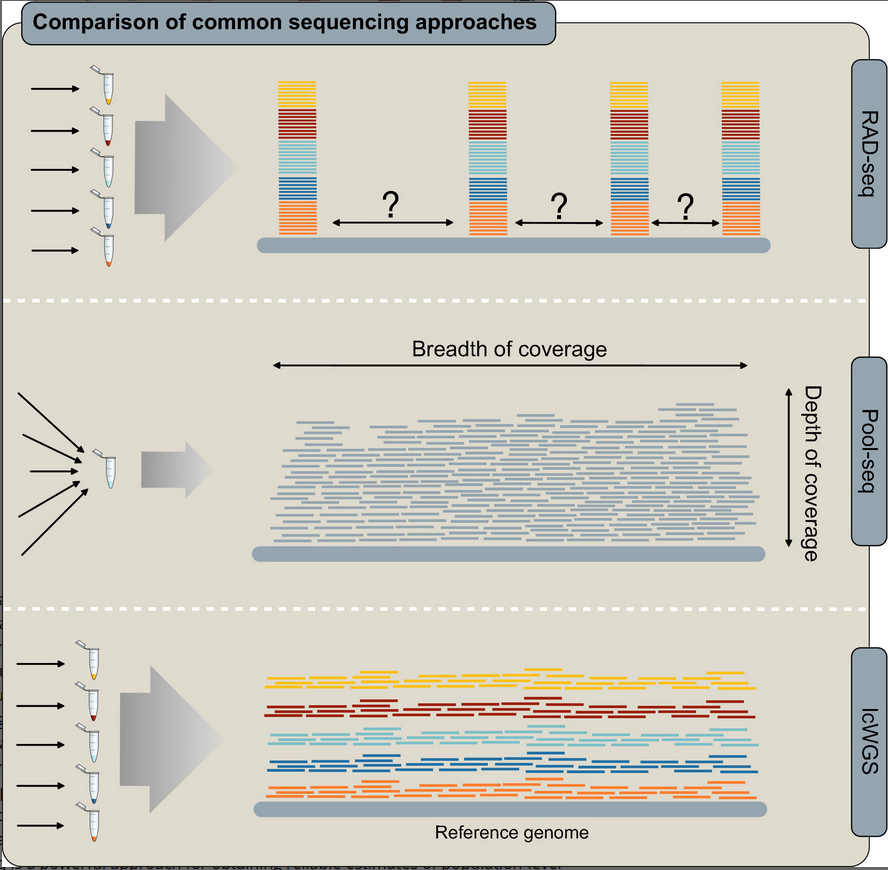
Il est préférable de séquencer a basse profondeur une population grande que de séquencer avec une bonne profondeur un nombre restrein d’individus, cela donne de meilleurs estimations des parametres de la population. selon cette étude une profondeur de 1X a 2X par locus par individus (ce qui agrandit la taille d’échantillon) maximise l’information obtenu sur la population.
A effort de sequencage équivalent (compromis entre la taille des samples et la couverture par sample) les meilleur résultats en therme d’estimation de fréquence d’allele, sont ce qui implique beaucoup de samples et donc pas beaucoup de coverage.
Objectifs
l’objectif principal du low-pass est de proposer un séquencage a très bas cout tout en ayant une information suffisante pour produire une analyse: - Analyse au niveau de la population: - calcul de fréquence allélique - différentiation génétique entre population (FST) - détection des pression de sélections - Analyse au niveau de l’individu - Consanguinité (inbreeding) - Admixiture (découverte d’ancetre issu de population différentes) - Assignation a une population
Ainsi le low-pass permet de séquencer un grand nombre d’individus et avoir une couverture sur tous le génome.
Limites
Les méthode d’imputation de génotype en low-pass sont limité quand il sagit de traiter des donnée d’espece “nonmodel” car la plupart des outils d’imputation se base sur des panels de référence d’haplotype. Cependant il existe des méthode qui outrepasse ce probleme en utilisant une grande quantitée de sample (n>2000).
Avec un séquencage basse profondeur ne permet pas d’inférer correctement les génotypes individuel. Mais dans le cadre d’étude de population l’information individuelle n’a pas trop d’intérets, c’est les charactéristique de la population overall qui importe ( fréquence allélique, DL, patterns…).
l’utilisation d’un systeme de Probabilistic Analysis Framework permet d’obtenir des résultats plus robuste quand les donnée sont bruité au niveau de la population. A l’echelle de l’individu cela permet d’obtenir un signal exploitable (pour calculer une Consanguinitée ou une amixiture) meme s’il est composé de SNP incertain.
Glossaire
Fst (Fixation index)
C’est une mesure qui compare des fréquence d’allele entre des population. Plus la Fst est petite plus la fréquence allélique est similaire entre les populations.
Probabilistic Analysis Framework (prise en compte de l’insertitude)
Cela consiste a considérer tous les génotype possible en leurs associant des probabilitée ce qui transforme une donnée “AG” en : \(P(AA)=0.2 , P(AG)=0.6 , P(GG)=0.2\) Cela néssécite des outils adapter car habituellement l’insertitude de génotypage n’est pas prise en compte par les outils classique de génotypage.
LD (linkage disequilibrium)
le LD ou déséquilibre de liaison est la quantification de la non-indépendance statistique des fréquence d’allele \(P_A P_B \ne P_{AB}\). Le LD est calculer par paire de loci
Benchmarking imputation accuracy in the presence or absence of a reference panel
Ils ont utiliser des individus séquencer a grande profondeurs pour générer un panel de 502 individus (issu de plusieurs population différente, mais meme espéce). Un des objectif est de tester des méthodes d’imputation de génotypes sur des donnée lcWGS avec et sans panel de référence.
Pour estimer la précision de chaque méthode ils on utilisé 32 individus séquencer en low coverage mais aussi en high coverage pour savoir si l’imputation étais correcte ou pas.
Without panel STITCH
Pour une imputation sans panel de références ils on tester plusieurs méthode d’échantillonage: - randomly - Par familles
L’imputation a tendance a étre meilleure si les individus ne sont pas regrouper par famille mais plutot échantilloner aléatoirement.
With panel GLIMPSE
Methode
L’algorithme GLIMPSE Permet le phasage et l’imputation de génotype en 4 étapes clés:
- Chunking: on découpes les différents chromosomes en chunk pour que les phase de phasing et d’imputation soit plus efficasse.
- Phasing and imputation: GLIMPSE utilise une méthode itérative pour cette étape.
- Ligation: on reconstruit les chromosome a partir des chunk, sans perdre l’information de phasage
- Haplotype identification.
Il est important de noté que la présence d’un panel
Overall performence
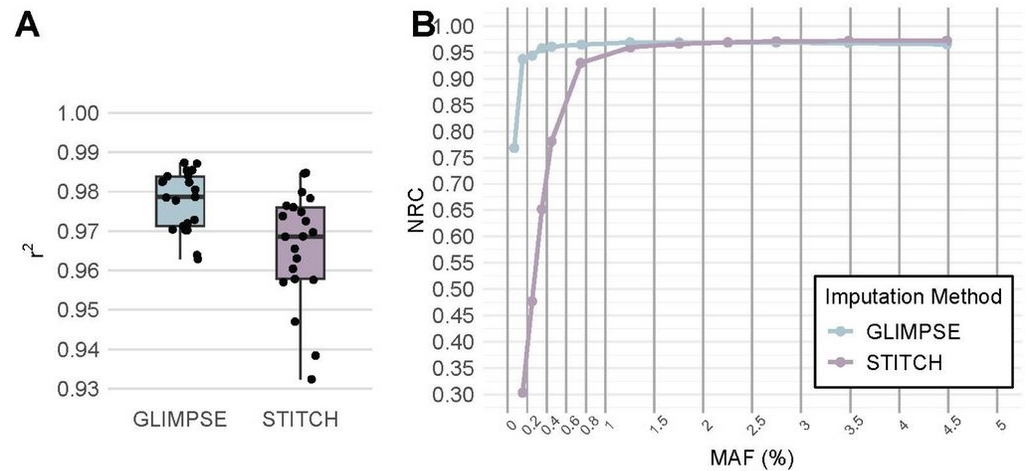
Les graphique ci-dessus représent l’efficassité des imputation de génotype, pour la figure A la métrique mesurée est la correlation de Pearson au carré \(r^2\). Calculé entre les donnée séquencer en haute qualitée (référence) et les donnée imputé.
On remarque que les résultats d’imputation son bon \(r^2>0.95\). La différence entre GLIMPSE et STITCH s’explique par le fait que GLIMPSE utilise une information suplémentaire qui est le panel de référence.
L’imputation utilisant un Panel de référence est significativement meilleure, en therme de moyenne et d’écart type que son homologue sans panel.
Comment obtenir le Panel
Pour obtenir le Panel de référence d’haplotype il faut génotyper avec une pipeline de découverte de variants. cela demande un séquencage assez précis.