Réunion 11/03/26
Lionel Viguier
Ordre du jours
- Pruning v2
- Bug de vraisemblance
Pruning v2
En l’état l’algorithme de pruning se divise en deux parties:
Calul des distance
Une partie de calcul des distance qui contient deux parcour du pédigree et les stocke dans deux dictionnaire différents:
distDict_gd stocke les distances des individus a leur descendant génotypé le plus proche. \[ \newline Dist_g = 0 | Dist_k = min(Dist_{Children_k})+1 \]
distDict_ga stocke les distances des individus a leur ancetre génotypé le plus proche. \[ \newline Dist_g = 0 | Dist_k = min(Dist(Mother_k), Dist(Father_k))+1 \]
Pruning v2
Filtre et suppression
Une partie qui vas filtrer et supprimer les noeuds indésirables dans le pédigree
Un individu est gardé si sa distance à un ancetre génotypé ET sa distance à un déscendant génotypé n’excéde pas une valeur seuil passé en parametres
le cas échéant il est gardé si il posséde un nombre d’enfants génotypé supérieur a 10
Pruning v2
def prunning(ped, gSamples, filter: int = np.inf):
allSamples = list(ped.nodes.keys())
distDict_gd = defaultdict( # gd for genotyped descendant
lambda: np.inf
) # initialise distance from a genotyped indiv to +inf
distDict_ga = defaultdict( # gd for genotyped ancestor
lambda: np.inf
) # initialise distance from a genotyped indiv to +inf
# set genotyped individuals distance to 0
for indiv in gSamples:
distDict_gd[indiv] = 0
distDict_ga[indiv] = 0
sortedPedNodes = sorted(list(ped.nodes.values()), key=lambda x: x.gen, reverse=True)
# crossing pedgraph generation by generation. First time we calculate distances
for i in sortedPedNodes:
indiv = i.indiv
if distDict_gd[indiv] != 0:
if i.children != []:
distDict_gd[indiv] = min([distDict_gd[c.indiv] for c in i.children]) + 1
sortedPedNodes = sorted(
list(ped.nodes.values()), key=lambda x: x.gen, reverse=False
)
# crossing pedgraph generation by generation. First time we calculate distances
for i in sortedPedNodes:
indiv = i.indiv
if distDict_gd[indiv] != 0:
if i.mother: # mean Father isn't None too
distDict_ga[indiv] = (
min([distDict_ga[i.mother.indiv], distDict_ga[i.father.indiv]]) + 1
)
# Removing individuals with a distance above filter
removed_cpt = 0
removed_indiv = []
for i in ped:
indiv = i.indiv
if distDict_ga[indiv] >= filter or distDict_gd[indiv] >= filter:
if len(i.children) < 10:
removed_indiv.append(ped.del_indiv(indiv))
removed_cpt += 1
else:
x = [j for j in i.children if distDict_gd[j.indiv] == 0 ]
# we dont prune individuals with at least 10 genotyped childrens
if len(x)< 10:
removed_indiv.append(ped.del_indiv(indiv))
removed_cpt += 1
print("Pruning done:", removed_cpt, "nodes removed")
return distDict_gd,distDict_gaBug de vraisemblance
Scaling
Nous avons décider de scaler les valeurs de posterieur et antériors ( les pénétrance sont déja scale si elles sont extraites d’un .VCF)<- a faire
Posterior
def _PeelUp(self):
for fam in sorted(
self.d.ped.fam_peel.values(), key=lambda x: x.gen, reverse=True
):
post_unscale = self.Posterior(fam)
# faire de post_unscale un pointeur vers self.d.Pij[..., fam.idn, :]
# <- return shape (nLoci, nGenotype, 2)
self.d.Pij[..., fam.idn, :] = (
post_unscale - np.max(post_unscale, axis=1)[:, None, :]
)Scaling
Anterior
def _PeelDown(self):
for fam in sorted(self.d.ped.fam_peel.values(), key=lambda x: x.gen):
# compute parents peel information here because it's the same for all childs
peel_parent = self.get_couple_peelProb(fam)
for child in fam:
# print(f"Child : {child.indiv}")
self.d.anterior[..., self.d.idx_dict[child.indiv]] = self.Anterior(
child, peel_parent
)
# scaling
# Faire un pointeur vers self.d.anterior[..., self.d.idx_dict[child.indiv]] pour lisibilitée
self.d.anterior[..., self.d.idx_dict[child.indiv]] = (
self.d.anterior[..., self.d.idx_dict[child.indiv]]
- np.max(
self.d.anterior[..., self.d.idx_dict[child.indiv]], axis=1
)[:, None]
)Résultats
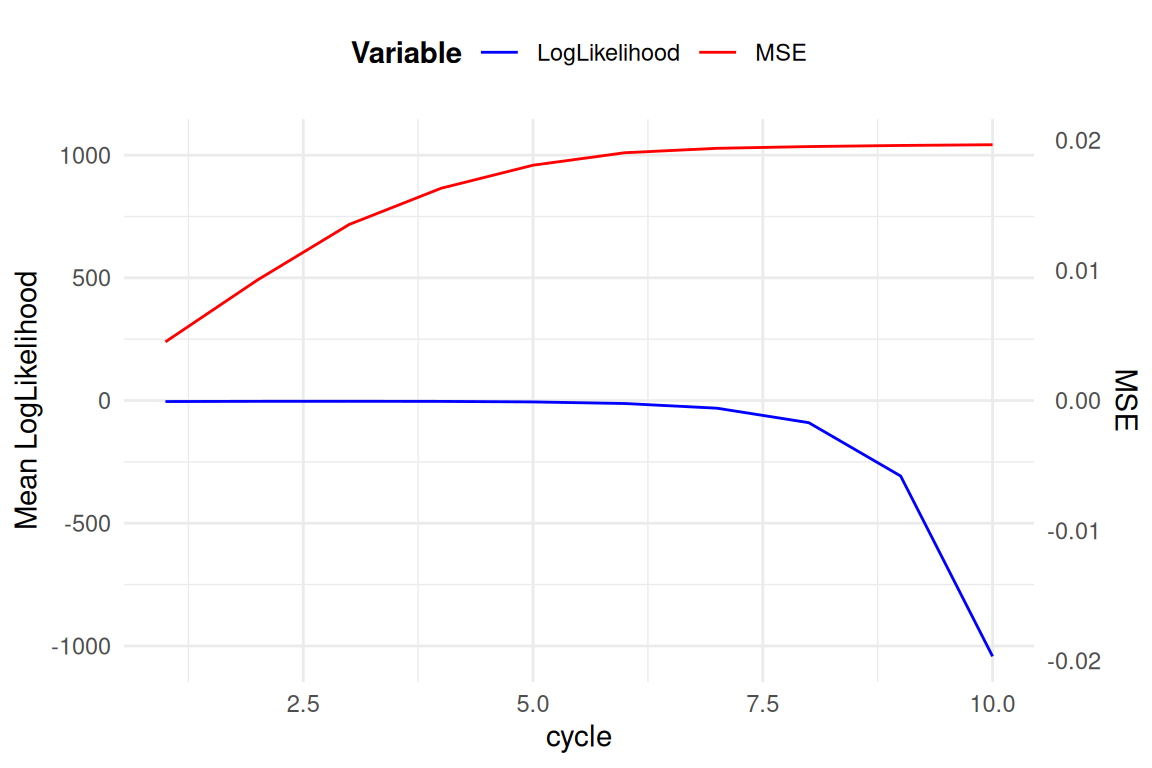
La MSE est calculé entre la “AAF” ou Alternative Allele Frequencies, réelle et estimée. Et la Log likelihood est L soit: la somme pour chaque locus et pour chaque individus, de la proportionelle a la probabilitée a postériorie de chaque génotype.
Anciens Résultats
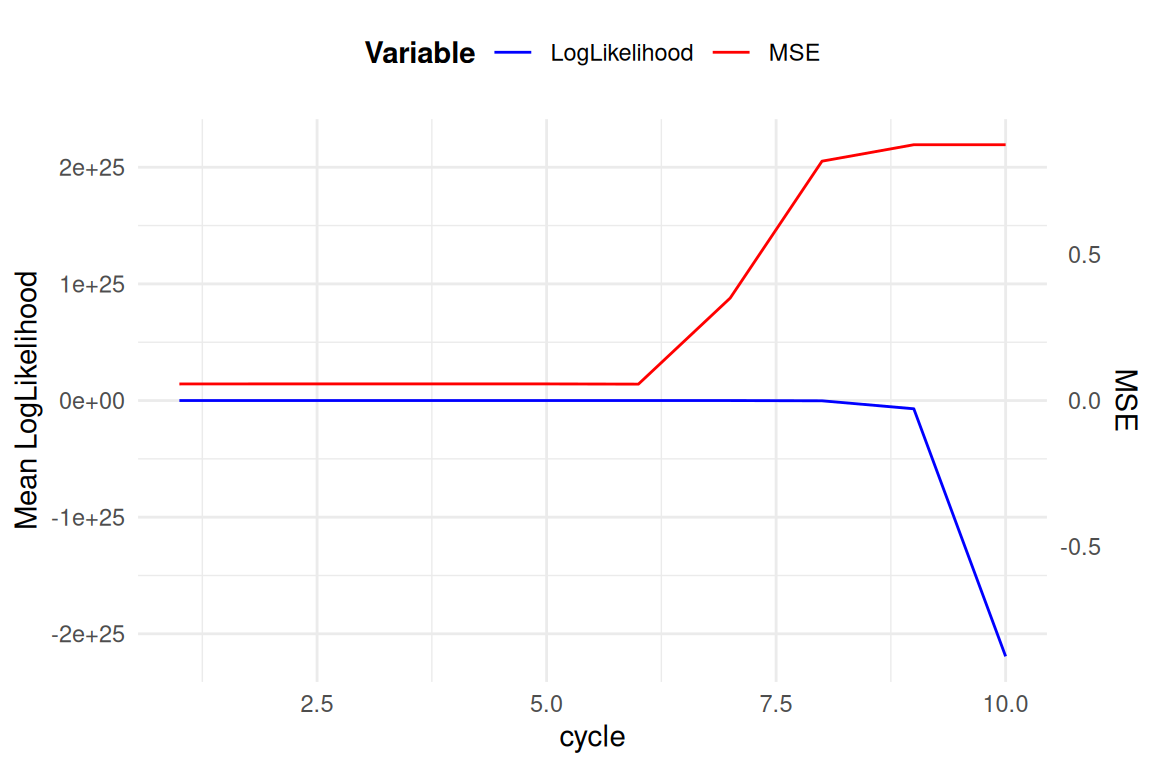
la concurence
Alphapeel
if operation == PEEL_DOWN:
for i in range(nOffspring):
child = family.offspring[i]
# Einstien sum notation 4: Project the parent genotypes down onto the child genotypes.
# anterior[child,:,:] = np.einsum("abci, abi -> ci", childSegTensor[i,:,:,:,:], parentsMinusChild[i,:,:,:])
projectParentGenotypes(
childSegTensor[i, :, :, :, :],
parentsMinusChild[i, :, :, :],
anterior[child, :, :],
)
anterior[child, :, :] /= np.sum(anterior[child, :, :], 0)Point hébdomadaire semaine 10.

Pied de page