Réunion 27/02/26
Lionel Viguier
Ordre du jour
- Pruning
- présentation jeux de donnée de Test
- Retour sur le calcul de la MAF chez les fondateurs
- Test et résultats
- Discussion
Pruning
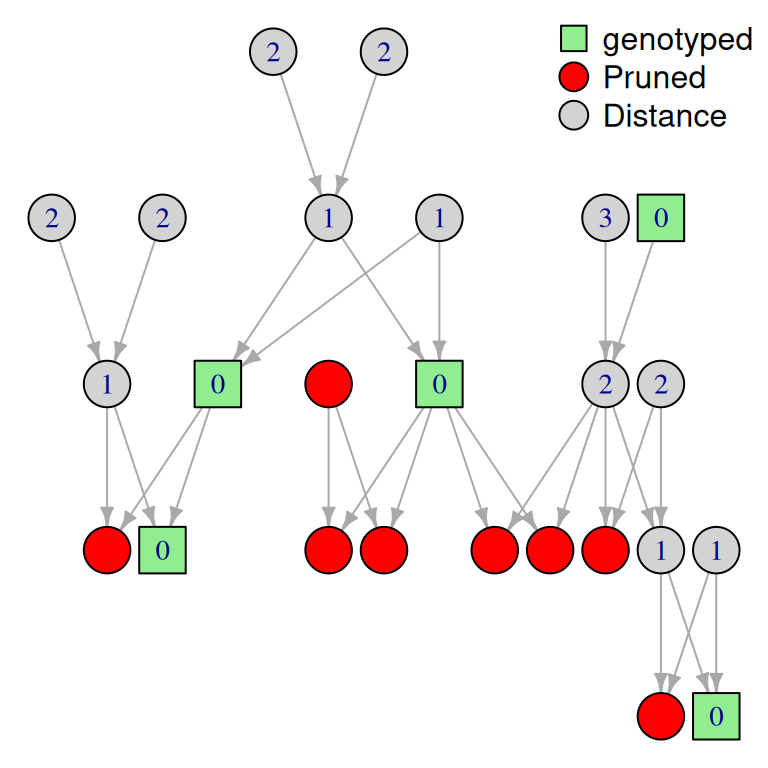
l’algorithme de pruning se base sur un tableau de distance au déscendent génotypé le plus proche:
- si un individu i est génotypé: \(Dist_i = 0\)
- si un individus i n’est pas génotypé: \(Dist_i = min(Dist_{childrens(i)}) +1\)
Présentation du JDD simulé
- Un fichier .fam avec un pédigree:
- 550 individus
- 11 générations (50 individus/génération)
- pas de saut de génération
- Un fichier de génotype réel:
- 1000 locus x 550 individues
- Un fichier .vcf de génotypage simulé:
- avec la Pénétrance
- Un fichier Pickle avec la MAF des fondateurs pour tous les loci
PopFreq
\[ \widehat{MAF} = {1\over 2} P(X = 1|Y) + P(X =2|Y) \]
def ReEstimateFoundersAnterior(samplesProbalog):
samplesProba = np.exp(samplesProbalog)
# shape (nloci,nGenotype,nfounders)
# Estimation of MAF
## we apply coefficient based on numbers of alt alleles
estimateMAF = samplesProba * np.array([0, 0.5, 1])[None, :, None]
estimateMAF = np.sum(estimateMAF, axis=(1, 2)) / samplesProba.shape[-1]
estimateMAF = np.log(estimateMAF)
log_1_minus_MAF = np.log(1 - np.exp(estimateMAF.clip(max=np.log(1 - 1e-10))))
# Reestimate founders anterior probabilities
reestimated_anterior_freq = np.zeros_like(
samplesProbalog[..., 0, None]
) # <- shape(nloci,nGenotype,1)
reestimated_anterior_freq[:, 0, 0] = 2 * log_1_minus_MAF # log((1-MAF)^2)
reestimated_anterior_freq[:, 1, 0] = (
np.log(2) + estimateMAF + log_1_minus_MAF
) # log(2 * MAF * (1-MAF))
reestimated_anterior_freq[:, 2, 0] = 2 * estimateMAF # log(MAF^2)
return reestimated_anterior_freqTest et Résultats
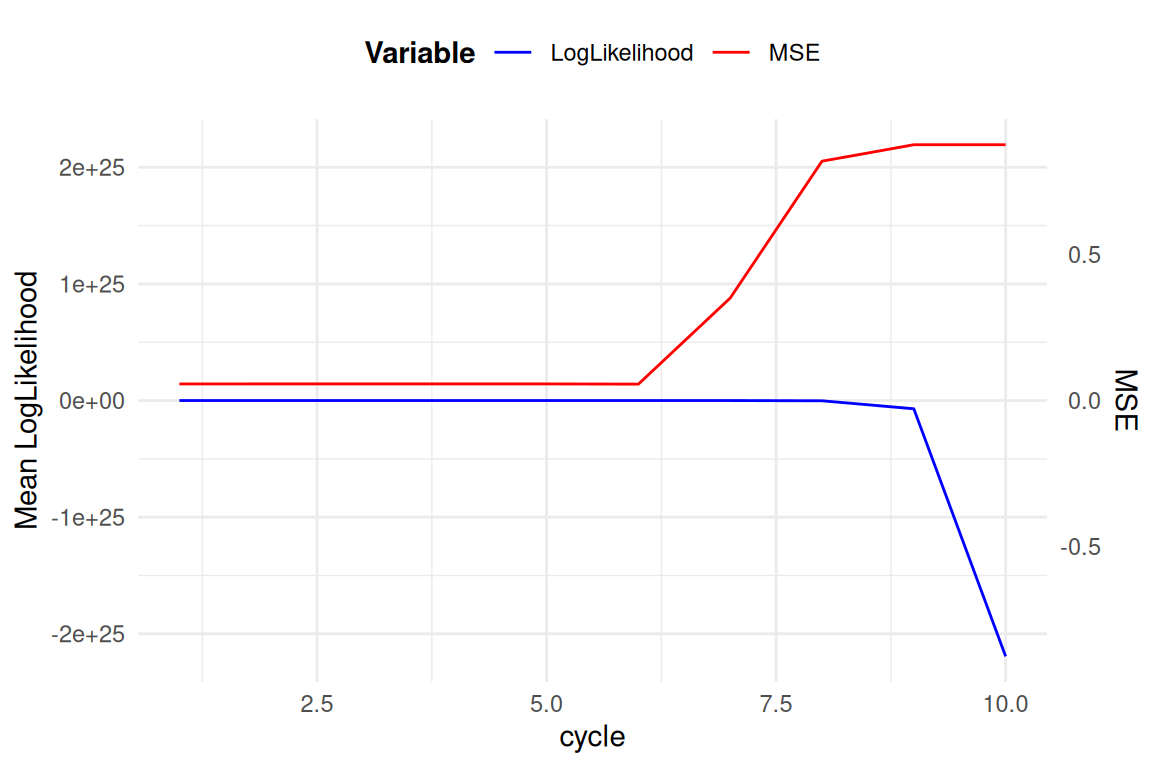
Sur le jeux de donnée simulé (avec conservation de tous les génotypages) on a pu traquer lors des différent cycles du peeling itératif
- la logvraisemblence L (résumé ici en moyenne sur les locus + individus)
- la \(MSE\) entre \(\widehat{MAF}\) et \(MAF\)
Discussion
- comment faire évoluer le prunning (en discuter ave Pierre et Vincent)
- dans le lowpass es ce que des individus serons génotyper en 30X?
- comment modifier un .vcf (pour faire des test en supprimant des génotypages)
- aspect pratique > quel package/commande utiliser?
- aspect théorique > y-a-t’il des animaux plus interessant a enlever du vcf que d’autres?
- résultats:
- la likelihood peut elle augmentée?
- es-ce-que une MSE a 5% est satisfaisante?
Retour
- Pruning:
- faire un vecteur de distance dans l’autre sens aussi (distance a l’ancetre génotyper le plus proche)
- compter le nombre d’enfants génotypé (filtre >10)
- ne pas faire de “in” mais utiliser un default dict
- Popfreq: Ne pas utiliser la fonction ReestimateAnterior avec la penetrance >>> faire du calling puis calculer avec les génotypes.
- Test et Resultats:
- Pensé a vérifier que les proba somme 1
- vérifier que les L sont égaux pour chaque individus a un locus donné
Point hébdomadaire semaine 8.

Pied de page